X fragile: il primo studio con gli organoidi cerebrali
Sono targati Sapienza e Iit i primi organoidi cerebrali 3d basati su cellule umane per lo studio dettagliato della sindrome dell’X fragile, una patologia genetica che si colloca fra le prime cause note di autismo e ritardo cognitivo nell’infanzia
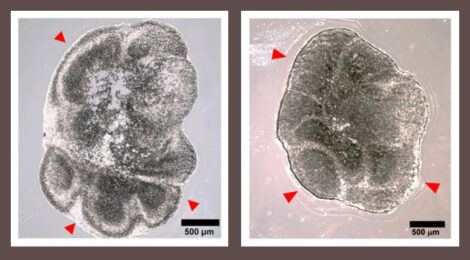
Disabilità cognitiva, ipereccitabilità sensoriale, problemi di linguaggio, crisi epilettiche: sono questi alcuni dei sintomi associati alla Sindrome dell’X fragile. Si tratta di una patologia rara – la sua incidenza si attesta intorno a 1/5000 nati – ma è fra le principali cause genetiche di autismo e ritardo cognitivo nell’infanzia. Una cura non c’è, non ancora. Un nuovo passo verso la comprensione della patologia e lo sviluppo di nuovi farmaci è stato fatto grazie alla costruzione dei primi organoidi cerebrali 3D – aggregati di cellule che assumono spontaneamente una precisa conformazione tridimensionale, finendo con l’assomigliare a organi in miniatura. I risultati sono pubblicati su Cell Death and Disease.

Cominciamo dal nome. Si chiama X fragile perché, in presenza della mutazione completa, il tratto terminale del cromosoma X sembra quasi rotto. La sindrome riguarda un gene (Fmrp) collocato appunto sul cromosoma X, che codifica una proteina (Fmr1) in grado di instaurare legami con l’Rna soprattutto a livello dei testicoli e del cervello, i tessuti più colpiti dalla sindrome. In condizioni patologiche, il gene ha una replicazione eccessiva di una tripletta nucleotidica: se tale replicazione supera le 200 volte – in condizioni normali non si eccedono le 50 – la proteina non viene prodotta. La sindrome interessa maggiormente individui di sesso maschile, poiché essi dispongono di un solo cromosoma X e non possono compensare la produzione della proteina con un secondo cromosoma.
Studi di laboratorio e analisi cliniche sugli animali non hanno avuto, finora, alcun successo a livello clinico – anche per le differenze fra i topi e l’essere umano. In questo contesto, lo sviluppo di organoidi 3D basati su cellule umane per replicare in vitro le strutture del sistema nervoso centrale è una novità.
“Ritengo che la sperimentazione animale sia imprescindibile, specie per la produzione di nuovi farmaci: confinare uno studio a un sistema in coltura non tiene conto dell’interazione fra i vari organi” spiega a StaR Silvia Di Angelantonio, professoressa associata al Dipartimento di Fisiologia e Farmacologia della Sapienza e all’Istituto Italiano di Tecnologia (Iit), e coordinatrice dello studio assieme a Alessandro Rosa del Dipartimento di Biologia e Biotecnologie della Sapienza. “Nell’ottica del principio delle 3R sulla sperimentazione animale – reduce, refine, replace – però, quello che possiamo fare grazie agli organoidi è ridurre il numero di animali necessari usando sistemi in coltura di derivazione umana, che consentano di arrivare a uno screening in vitro più avanzato di quello attuale.”
Un elemento fondamentale della tecnologia impiegata – precisa la ricercatrice – è l’utilizzo di cellule staminali di derivazione non embrionale. Si tratta delle cellule umane riprogrammate, una scoperta premiata con il Nobel nel 2012 che consente di prendere una cellula da un individuo (come un globulo bianco) e riportarla al suo grado di staminalità.
“Sono cellule con un grande potenziale e, rispetto alle staminali degli embrioni, hanno un vantaggio etico enorme” continua Di Angelantonio. “Poi, sono lo strumento ideale per la medicina personalizzata: si può studiare una malattia genetica su un singolo paziente e l’effetto di un farmaco sul suo sistema. Significa curare la persona, non la malattia.”
Nello specifico, sono state usate cellule di controllo sane e cellule dello stesso campione in cui il gene Fmrp è stato silenziato, in modo da operare in condizioni in cui l’unica differenza fosse l’assenza della proteina Fmr1. Ora i risultati: Si osserva, fra gli organoidi dei due campioni, uno squilibrio nello sviluppo dei neuroni rispetto ad altre cellule del cervello, come gli astrociti – responsabili del mantenimento di una corretta comunicazione nel sistema nervoso. Inoltre, si rileva ipereccitabilità nell’attività di rete delle cellule in cui manca la proteina, che gli autori associano alle crisi epilettiche dei pazienti X fragile.
“Ora stiamo studiando organoidi ottenuti da cellule di pazienti X fragile. È necessario capire come la patologia vera e propria influisce sullo sviluppo cerebrale, perché nessun paziente reale ha solamente un gene silente come nel nostro modello” conclude Di Angelantonio. “Grazie alla collaborazione con il gruppo di farmacologia dell’Iit di Genova, cominceremo anche a testare nuovi farmaci sugli organoidi.”
Crediti: Istituto Italiano di Tecnologia



















BUONGIORNO SONO LA MAMMA DI UN RAGAZZO DI 27 ANNI AFFETTO DA X FRAGILE
VORREI SAPERNE DI PIU